Медицинская реабилитация
Некоторые аспекты патофизиологии травматического повреждения и регенерации спинного мозга
И. А. Борщенко, А. В. Басков, А. Г. Коршунов, Ф. С. Сатанова
НИИ нейрохирургии им. акад. Н. Н. Бурденко (дир. - акад. РАМН А. Н. Коновалов) РАМН, Москва
Травматическое повреждение спинного мозга (СМ) является одним из актуальных вопросов современной медицины. Высокая частота позвоночно-спинальной травмы сочетается со сложностью патогенеза травматической болезни СМ и недостаточной эффективностью методов лечения. Практически полная инвалидность больного и огромные затраты на лечение и реабилитацию выводят эту проблему за рамки чисто медицинских аспектов [4, 8, 11, 13]. Попытки перенести принципы восстановления периферической нервной системы на СМ без учета особенностей его морфологии и физиологии не привели к успеху [7]. Механическое сопоставление поврежденных отрезков СМ, эмпирическое применение различных методов воздействия на поврежденную ткань оказалось неэффективным для восстановления функции этого органа [1, 2, 5, 9, 11]. Только после изучения ультраструктурных, молекулярно-клеточных механизмов повреждения стали понятны некоторые причины неудач и наметились дальнейшие пути исследования.
Постоянно растущее число работ, посвященных проблемам травмы и регенерации СМ, заставляет еще раз взглянуть и оценить состояние проблемы на сегодняшний день. Прогресс в исследованиях патогенеза повреждения СМ можно объяснить прежде всего успехами в развитии методик молекулярной биологии, биохимии. Современные иммуногистохимические методы позволяют получить антитела практически к любому клеточному метаболиту, а способы визуализации микроскопических объектов дают возможность наблюдать четкие картины их распределения. Кроме того, появившиеся методы воздействия на клетку - использование генной терапии в виде вирусных агентов, катионных липосом с генным материалом - стимулируют дальнейшие исследования в этой области [25].
Травматическое повреждение СМ не ограничивается локальным разрушением структур, но запускает серию связанных в пространственно-временном отношении реакций, приводящих к вторичной гибели изначально неповрежденных нейронов и глии.
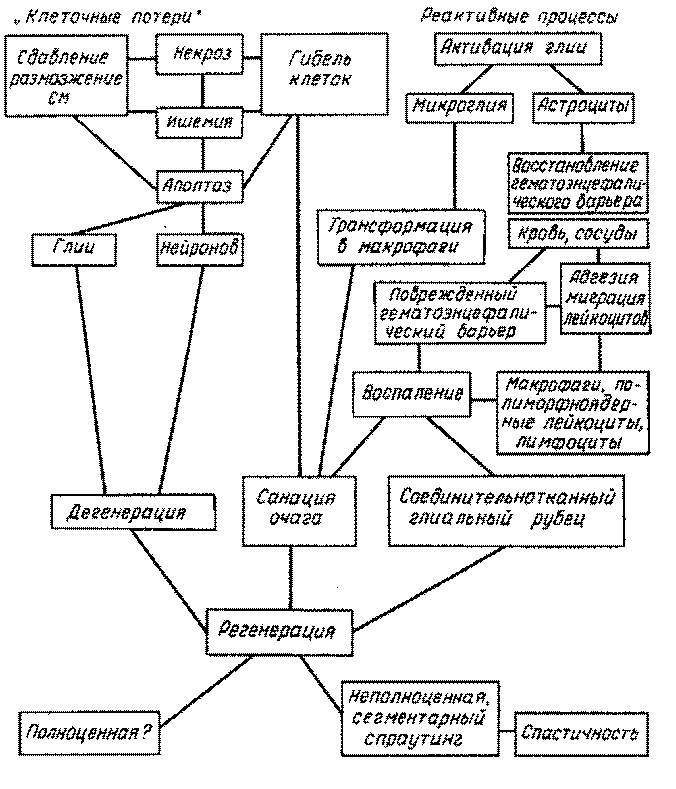
Проявляется это в виде гибели нейронов, восходящей и нисходящей дегенерации нервных волокон и в конечном итоге отсутствием полноценной регенерации в зоне травмы. Вторичные патологические изменения после первичной механической травмы включают петехиальные кровоизлияния, прогрессирующие до геморрагического некроза, энзиматический липидный гидролиз с продукцией эйкозаноидов, свободнорадикальноелипидное окисление, инфлюкс Са2+, увеличение протеазной активности, накопление возбуждающих аминокислот, кининов, серотонина, динорфина, ишемию с результатом в виде уменьшения напряжения кислорода в тканях и воспалительным нейронофагоцитозом полиморфноядерными лейкоцитами [59]. Множественность и комплексность процессов, следующих за травматическим повреждением, затрудняет оценку роли специфического механизма во вторичном повреждении [57]. Рассмотрим условную последовательность патофизиологических клеточных реакций, которые запускаются в момент травматического повреждения, но максимально проявляются в разные временные периоды. Наиболее ранним является воспалительный ответ.
Воспаление. Механизм воспаления, будучи универсальным клеточным ответом организма с включением его иммунного и неиммунного компонентов, является ключевой реакцией в посттравматическом процессе. Хотя воспаление является главным механизмом в санации очага повреждения, избыточный воспалительный ответ может привести к вторичному повреждению путем избыточного выделения медиаторов воспаления и развития гиперергических клеточных реакций, что в свою очередь вызывает и усиливает такие процессы, как ишемия и апоптоз. Уже через 24 ч наблюдается максимальная инфильтрация области травмы полиморфноядерными лейкоцитами, через 24- 48 ч - пик миграции макрофагов, через 48 ч - натуральных киллеров, хелперов и супрессоров. Последние упомянутые клетки, участвующие в иммунной модуляции воспаления, наблюдаются до 16 сут. [56]. Лейкоциты, появляющиеся в очаге травмы, выделяют множество прямых цитотоксичных факторов и медиаторов воспаления, позволяющих процессу воспаления самоподдерживаться и расширяться вне очага поражения. In vitro установлено вторичное повреждение путем выделения миелопероксидазы полиморфноядерными лейкоцитами [28], а назначение клодроната, подавляющего макрофаги, увеличивает сохраняемость миелинизированных трактов [61]. Блокирование выработки мононуклеарными фагоцитами токсичной квинолиновой кислоты (продукт обмена триптофана) уменьшает неврологический дефицит in vivo [53]. Макрофаги, микроглия участвуют в прогрессирующем некрозе путем освобождения свободных радикалов и воспалительных цитокинов - интерлейкина-1, интерлейкина-6, туморнекротизирующего фактора, факторов адгезии тромбоцитов (IL-1, IL-1b, TNFа, PAF соответственно) [27, 56]. Медиаторы воспаления имеют множество мишеней и как результат - развитие многоуровневого воспалительного иммунного ответа. IL-1a выделяется микроглией, макрофагами и стимулирует in vitro выработку в глиальных клетках трансформирующего фактора роста, увеличивает отек астроцитов, вызывает гипертрофию растущих астроцитов [51]. Поврежденный эндотелий сосудов так же выделяет IL-1a, IL-1b, TNFа; травма индуцирует образование внутриклеточных молекул адгезии (IСАМI), являющихся иммуноглобулинами [42, 56]. Система гемостаза с первых мгновений повреждения участвует в процессе воспаления и начальной его фазе - адгезии лейкоцитов. Тромбин вызывает пролиферацию гладкомышечных, эндотелиальных клеток, увеличивает продукцию эндотелина, усиливает адгезию лейкоцитов; назначение гирудина, как антагониста тромбина, уменьшает воспалительную клеточную инвазию [54]. Пристальное внимание уделяется таким межклеточным посредникам, как интерлейкины. IL-1 через рецепторы глутамата может вызывать гибель клетки путем апоптоза; IL-6 участвует в регенерации (стимулирует выработку антител В-лимфоцитами, повышает Т-цитотоксичность); IL-10 является противовоспалительным, ограничительным звеном в цепи воспалительных реакций (уменьшает выработку IL-1, IL-6, IL-8, TNFа, INF) [77]. IL-1b усиливает кровоток в СМ [62, 72].
Такое множество медиаторов воспаления, их прямых и обратных связей с различными клетками дает широкую возможность воздействия на травматический процесс. Назначение антител к IL-1, TNFа уменьшает отек, снижает нейрональную смерть, микроглиальную пролиферацию. Блокада освобождения или экспрессии медиаторов воспаления после прохождения гематоглиального барьера лейкоцитами либо торможение адгезии и миграции лейкоцитов в очаг воспаления (через воздействие на систему гемостаза, молекулы адгезии) - вот возможные пути влияния на воспалительный компонент травматического процесса [56].
Одновременно с воспалительной реакцией развивается и глиальный ответ.
Глиальная реакция. Глия как клетки, создающие особое окружение нейронов, играет важную роль в процессе санации очага травмы и обеспечивает возможность нейронального выживания и восстановления. Уже через 24 ч в зоне травмы с участием системы комплемента активируются микроглиоциты, которые превращаются в макрофаги, активно поглощающие детрит. В дальнейшем комплементнезависимым путем микроглия активируется вдоль проводящих пучков на отдалении от места травмы, участвуя в процессе вторичной дегенерации волокон [76]. Микроглиальные клетки производят отростки, контактирующие с олигодендроцитами, и путем прямого фагоцитоза или выделения цитокинов (TNF) уничтожают миелин в проксимальном и дистальном отрезках мозга (в отношении к месту травмы) [69]. Несмотря на общую активацию глиальных клеток, в них усиливаются процессы программированной клеточной смерти - апоптоза, что во многом определяет дальнейшее развитие дегенеративного процесса. И прежде всего апоптозу подвергаются клетки, тесно контактирующие с аксоном. Апоптоз олигодендроцитов приводит к быстрому набуханию миелина и заключению оставшихся олигодендроцитов как бы в изоляты, ловушки. Возможно, смерть этих интерфасцикулярных клеток является необходимой подготовкой для регенерации [17]. На наиболее ранних стадиях глиального ответа - 1-3 дня после травмы - реагируют астроциты как компонент гематоспинномозгового барьера. При этом клетки перестраиваются на ранние стадии онтогенетического функционирования, что подтверждается продукцией виментина, глиального фибриллярного кислого протеина - белков быстроразвивающихся астроцитов [22].
Тот факт, что травматическое повреждение не просто разрушает клеточные структуры путем травматического, ишемического некроза, но глобально изменяет всю жизнедеятельность сохранившихся клеток, подтверждается развитием отсроченной программированной гибели клеток - апоптозом.
Апоптоз. Гибель клетки путем апоптоза - есть включение травматическим агентом механизмов естественной клеточной смерти. Причиной развития апоптоза может быть прямое воздействие на геном клетки (вирусы) или непрямое влияние через нейромедиаторы (глутамат), медиаторы воспаления, ишемию и пр. Такая полиэтиологичность апоптоза связывает его со многими патологическими состояниями - травмой, ишемией, инфекцией. Современные методы позволяют выявить самые ранние стадии апоптоза в клетках травмированного СМ. При этом механизм травмы - сдавление мозга в эксперименте (drop-weight model) или его пересечение - не имеет значения. Такие методики как TUNEL, ISEL позволяют выявить изменения ДНК на самых ранних стадиях апоптоза, до того как появятся тельца апоптоза - фрагментированная ДНК в виде глыбок хроматина в ядре или в цитоплазменных каплях вне клетки [66]. Процессы, связанные с апоптозом, наблюдаются уже спустя 6 ч после травмы. Первый пик гибели клеток происходит примерно через 3 дня - апоптозу подвергаются как нейроны, микроглия, так и в меньшей степени олигодендроглия; второй пик - это максимальная гибель олигодендроцитов к концу 2-й недели. Предполагают, что глиальный апоптоз может быть причиной аксональной дегенерации [29, 79]. Изменения ДНК, сопровождающие апоптоз, проявляются экспрессией особых генов и трансляцией им соответствующих белков. Такие агенты, как Са2+, глутамат, ишемия, гипоксия, запускают включение ранних немедленных генов с продукцией соответствующих белков, в частности теплового шокового протеина, белка р53. Последний, известный как белок-супрессор опухолей, участвует в восстановлении ДНК поврежденной клетки. Максимум его содержания находят через 2 сут; исчезает к 7-м суткам постгравматического периода. Обнаруживается р53 в глиальных клетках и на некотором удалении от места травмы СМ - и это является ранним ответом, предшествующим ваалеровской дегенерации волокон [66, 68]. Конечная фрагментация хроматина связана с активацией эндонуклеаз, расщепляющих ДНК. Параллельно происходит оксидация белка ДНК, маркером которой является накопление протеинкарбонилов [49].
Таким образом, апоптоз нейронов приводит к прогрессирующей потере числа активных клеток, а апоптоз глии препятствует выживанию и прорастанию оставшихся волокон, что выражается в отсутствии полноценной регенерации в СМ.
Из наиболее явных опосредованных механизмов травматического повреждения можно выделить ишемию.
Ишемические нарушения. Ишемия СМ является непременным компонентом его травматического повреждения. Простое сдавление сосудов СМ без травматического повреждения клеток приводит к ишемическому некрозу, развитию воспалительной реакции, запускает апоптоз. Уже через 180 мин компрессии сосудов мозга наступают полностью необратимые изменения нейронов [26]. Травма и ишемия вместе ответственны за клиническое проявление в виде пара-, тетраплегии из-за повреждения белого вещества СМ. Хотя детали молекулярного каскада, ведущего к необратимой аксональной дисфункции в СМ при спинномозговой травме и ишемии только частично изучены и понятны, эти результаты могут иметь практическое применение в отношении этих нарушений. При этом обсуждается роль Nа+/Са2+-, Nа+/Н+-насосов, возбуждающих аминокислот (глутамат, аспартат), ионов Са2+ в патогенезе ишемических процессов, связанных с травмой СМ [41, 50, 66]. Ишемия приводит к повреждению гематоспинномозгового барьера - снижается содержание специальных молекул барьера - глюкозо-1-транспортер (GLUT-1) и эндотелиально связанного антигена [43].
Именно воздействие на ишемический компонент травмы является наиболее доступным на настоящий момент как медикаментозными способами (изменением реологических свойств крови, влиянием на свертывающую систему, на тонус сосудов), так и хирургическим путем (декомпрессия мозга).
Описанные клеточные реакции на субклеточном уровне реализуются путем активации биологически активных веществ, которые изменяют как функционирование клеток, так и внутриклеточную структуру. В свою очередь первичные структурные повреждения вызывают освобождение ранее неактивных веществ, которые приводят к вторичному повреждению структуры клеток. Такая связь между структурой и функцией лежит в основе формирования пространственной и временной цепи взаимосвязанных первичных и вторичных патологических реакций.
Рассмотрим некоторые субклеточные и молекулярные механизмы вторичной травмы.
Повреждение цитоскелета. Оценивая все более тонкие уровни повреждения при растяжении аксонов, наблюдают прямое разрушение их мембраны - аксолеммы, прямое и отсроченное разрушение цитоскелета. Растяжение приводит к непосредственному повреждению аксональных микротрубочек, микрофиламентов [52, 72]. Количественный анализ острой аксональной патологии при экспериментальной контузии СМ показал общую тенденцию в повреждении СМ: в течение 15 мин повреждается серое вещество, в течение 4 ч - белое. Медленное развитие повреждения белого вещества дает терапевтическое "окно" для воздействия на процесс. Уже через 15 мин 60% аксонов повреждается: наблюдается аксональный коллапс цитоскелета в виде отсутствия микротрубочек, плотного расположения нейрофиламентов. Это сочетается с развитием внутриклеточного ацидоза, накоплением внутриклеточного Са2+ (вовлекаются Nа+/ Са2+-, Nа+/Н+-насосы) [67]. Аксональная травма сопровождается протеолизом цитоскелета, и здесь первичным энзимом разрушения определен кальпейн, являющийся Са2+-активируемой протеазой. В результате протеолиза цитоскелета определяют уменьшение содержания нейрофиламентного протеина NF-200 и рост фрагментов деградации спектрина (субстрат кальпейна) [21, 70. 78].
Роль ионов Са2+, К+, Nа+. Стало бесспорным, что изменения в Са2+-обмене играют роль в патофизиологическом каскаде клеточных изменений, которые ведут к нейрональной смерти и дегенерации после травмы ЦНС. Изменения в гомеостазе ионов Са2+ лежат в основе клеточной смерти при ишемии СМ. Отмеченные изменения в концентрации ионов Са2+ в зоне травмы линейно коррелируют с размером нанесенной травмы СМ в эксперименте [56]. Са2+ является одним из вторичных месенджеров между мембраной и клеточными ферментными системами, между мембраной и генным аппаратом. Отмечена экспрессия некоторых генов при достижении концентрации Са2+ определенного уровня. Повышение внутриклеточного Са2+ приводит к абсорбции его митохондриальными мембранами и последующим блокированием дыхательной цепи электронов. Повышенное внутриклеточное содержание Са2+ активирует нелизосомальную цистеиновую протеазу кальпейн, приводя к лизису цитоскелета, деградации энзимов (киназ, фосфолипаз), мембрано-ассоциированных белков (ионных каналов, переносчиков, рецепторов, молекул адгезии). Расположенный и в нейронах, и в глии, кальпейн оказывается вовлеченным в постишемическую и посттравматическую цитотоксическую реакцию, связанную с повышением внутриклеточного Са2+. Несмотря на большое число публикаций, вопрос о роли Са2+ при травме СМ остается открытым [20, 26, 41, 6б]. Исследуются также вопрос о роли Nа+/Са2+-каналов на выживание нейронов. В частности, указывается на открытие электровозбудимых каналов при ишемии с дальнейшим включением Nа+/Са2+-, Nа+/Н+-насосов, развитием внутриклеточного ацидоза и повышением внутриклеточного Са2+ [19, 36, 58, 73]. Отмечено, что хотя снижение внеклеточного Са2+ важно для выживания клеток, это может быть препятствием к новому росту аксонов [33].
Влияние нейромедиаторов. Особую роль в механизмах первичного и вторичного повреждения клеток уделяют возбуждающим медиаторным аминокислотам - глутамату и аспартату. На моделях животных доказана токсичность этих аминокислот в отношении нейронов. Избыточное содержание данных медиаторов при травме, ишемии может привести как к некрозу, так и к апоптозу клеток. Установлены некоторые механизмы их влияния на клетку: через инфлюкс Са2+ в клетку либо через лизис фосфатидилинозитола фосфолипазой С с освобождением Са2+ из внутриклеточных депо (эндоплазматической сети, митохондрий). Данные о влиянии возбуждающих аминокислот in vitro и in vivo имеют некоторые противоречия, и механизмы их действия спорны. Но какой бы механизм не был, доказательства на моделях животных их цитотоксичности указывают на их возможную ответственность за посттравматические эффекты, которые можно блокировать антагонистам рецепторов возбуждающих аминокислот [34, 56, 60, 80].
Свободнорадикальнос окисление, метаболиты арахидоновой кислоты. Исследуется связь рецепторов глутамата, аспартата, ионов Са2+ с образованием метаболитов арахидоновой кислоты. Посттравматическое изменение в содержании внутриклеточного Са2+ усиливает атаку на клеточные мембраны и цитоархитектуру путем активации кальпейна, липаз и индуцирует образование патологических элементов из каскада арахидоновой кислоты. Показано, что травма вызывает отсроченную продленную деградацию мембранных фосфолипидов [56]. Значительно повышается содержание инозитол-3-фосфата, что самостоятельно приводит к росту внутриклеточного Са2+ [35].
Посттравматические повреждения мембран и уменьшение доставки субстрата при ишемии могут участвовать в продукции метаболитов арахидоновой кислоты, активных форм кислорода (О*). Источник О* - освобождение его из митохондрий при повышении содержания Са2+, моноаминоксидазы, циклоксигеназа, синтетаза NO. Высокий уровень глутамата может генерировать высокий уровень О* после травмы ЦНС. Свободное реактивное железо также может участвовать в образовании О*. О* приводит к деструкции билипидных мембран, окислению протеинов, нуклеиновых кислот. О* может повреждать некоторые регуляторные процессы, включая цитокины и факторы роста, которые обусловливают включение генов, участвующих в апоптозе [31, 35, 48, 56, 63, 71]. Травма приводит к истощению эндогенных антиоксидантов - витаминов А, Е, С, убихинона, это способствует посттравматической клеточной смерти путем неконтролируемого перекисного повреждения, которое может распространяться в изначально неповрежденном мозге [56].
Перспективы регенерации. Ранее считавшийся минимальным, регенераторный потенциал СМ в настоящее время оценивается более оптимистично [3, 6, 7, 11]. Исследуются молекулярные условия и клеточные влияния на восстановление и рост волокон сквозь соединительнотканно-глиальный рубец. Пептидные нейротрофические факторы роста, включая фактор роста нервов, основной фактор роста фибробластов, цилиарный нейротрофический фактор, нейротрофический фактор головного мозга, инсулиноподобный фактор роста, нейротропин-3, функционируют в мозге для поддержания нейронального выживания, индуцируют спраутинг нейронов, обеспечивают направление роста нейронов [7, 10, 16, 23, 32, 44, 56, 74]. Травма изменяет соотношение и увеличивает число факторов роста, но в дальнейшем происходит их истощение [5б]. Это дает основание стимулировать аксональный рост введением этих факторов: прямым назначением либо трансплантацией фибробластов, продуцирующих факторы роста [39, 55, 56]. Факторы роста могут быть ответственны за избыточный посегментарный спраутинг, что приводит к дисфункциональному выздоровлению, гиперрефлексии, спастичности [44, 45]. В качестве трансплантируемых тканей к месту рассечения СМ рассматриваются фетальные клетки как замена погибших, аутонервы как направляющие структуры и триггер роста аксонов [6, 10, 12, 14, 40, 65, 81, 83]. Определяются клеточное окружение прорастающих аксонов и связь их с растущей глией. Используя фибробласты, продуцирующие ростовые факторы, добиваются прорастания аксонов сквозь глиальный рубец [7, 12, 38]. В связи с недостаточностью повторного прорастания волокон рассматривается гипотеза об иммунной привилегированности ЦНС млекопитающих и торможении регенерации развитой иммунной системой. Для его преодоления используют активированные макрофаги. Макрофаги как клетки, осуществляющие презентацию антигена иммунным клеткам, инкубируют с сегментом периферического нерва, после чего имплантируют к месту пересечения СМ. Такой активацией достигается уменьшение иммунного торможения роста и наблюдается прорастание волокон, что приводит к частичному восстановлению движений. Кроме того, прорастание доказано электрофизиологически с оценкой вызванных потенциалов после повторной перерезки выше первичной секции мозга [46, 47].
Таким образом, регенерация СМ с прорастанием аксонов сквозь соединительнотканно-глиальный рубец при определенных условиях возможна, и поиск этих условий и причин, препятствующих восстановлению функции, является главным направлением в исследованиях травматического повреждения СМ.
Поскольку описывается большое количество данных, указывающих на многие различные нейрохимические, клеточные и молекулярные каскады, каждый со своим уникальным временным включением, и вовлеченных в опосредованно посттравматических патофизиологических механизмов, приводящих к вторичному отсроченному повреждению мозга после травмы, вероятно, что рациональная во временном отношении терапевтическая стратегия, нацеленная как на ранние процессы повреждения (Са2+-, глутамат-, O*-опосредованные), так и на поздние хронические (воспаление, восстановление и регенерация), будет более эффективна, чем одна лечебная стратегия [56]. Представляется важным подчеркнуть, что предотвращение и преодоление вторичного повреждения может стать тем краеугольным камнем, который позволит сохранить и в дальнейшем восстановить структуру и функцию СМ.
Комментарий. Авторами представлен обзор литературы последних лет, посвященный некоторым аспектам патогенеза травматической болезни спинного мозга. Наряду с известными нейрохирургам первичными разрушениями структур спинного мозга, вторичными ишемическими и геморрагическими повреждениями, метаболическими и сосудистыми расстройствами описаны и другие патофизиологические изменения в поврежденном мозге. Отражено большое значение для вторичного повреждения травмированного мозга избыточного образования нейромедиаторов, медиаторов воспаления вследствие неуправляемости гиперергической реакции, эндонуклеаз, ионов кальция и других биологически активных веществ. Описаны некоторые причины апоптоза нейронов и глии, ведущих к прогрессированию дефицита функционально активных структур мозга.
Патофизиологические аспекты травматической болезни спинного мозга описаны авторами по результатам экспериментальных исследований зарубежных и отечественных ученых. Они базируются на современных достижениях молекулярной биологии и биохимии. Появилась возможность получать специфические антитела к различным клеточным метаболитам, маркировать их и определять количественно. Чрезвычайно важно, что наметились некоторые пути целенаправленного влияния на некоторые звенья патологических процессов, протекающих в травмированном мозге, с целью профилактики вторичных его повреждений. Известно, что злокачественность течения травматического повреждения мозга обусловлена не только выраженным магистральным типом его кровоснабжения, но и плохой управляемостью целого каскада патологических реакций, приводящих к расширению очага первичного повреждения мозга. Эти экспериментальные данные могут в ближайшее время получить выход в клинические исследования с целью применения в практике нейрохирургов.
Вызывают определенный оптимизм результаты экспериментов на животных по использованию фибробластов и активированных макрофагов для усиления прорастания аксонов поврежденного спинного мозга через глиальный рубец, а также трансплантации эмбриональной нервной ткани с целью восполнения клеточно-глиального дефицита и заместительного выделения биологически активных веществ.
Обзорная статья И. А. Борщенко и соавт. своевременна и полезна для стимулирования научного поиска нейрохирургов в сфере поиска средств, повышающих результаты лечения больных с травматической болезнью спинного мозга.
А. А. Луцик (Новокузнецк)